Delcath Systems Inc. (NSDQ:DCTH) CEO Eamonn Hobbs is in a good mood these days.
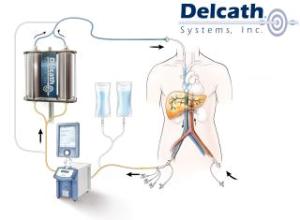
Despite some regulatory hiccups in the U.S., the regulatory picture overseas is rosier for Delcath’s proprietary focused chemotherapy delivery system after winning CE Mark approval in the European Union in April. As a consequence, Delcath is gearing up for the fourth-quarter European commercialization of the focused chemotherapy delivery system, Chemosat. The device uses a system of tubes to localize the delivery of toxic chemotherapy agents by isolating the targeted organ — in this case, the liver — from the rest of the body’s bloodstream.
One tube delivers melphalan hydrochloride to the liver via a catheter inserted through the femoral vein and positioned in the hepatic artery. A second tube draws blood mixed with the drug toward an external filtration system; a third delivers the now-filtered blood back to the heart via the internal jugular vein.
Isolating the drug allows physicians to use doses several times higher than current standards, and allows physicians to follow up a focused dose of chemo to the liver with a systemic dose to target cancerous tissue throughout the body.
Check out this animated demonstration of the Chemosat system
Hobbs claims the Chemosat system is a cancer treatment breakthrough that, although only indicated for liver cancer treatment so far, may have applications in other organs.
On this side of the pond, the FDA dealt Delcath a setback two months ahead of the CE Mark, issuing a Refusal to File letter on a new drug application for the Chemosat system.
Hobbs told MassDevice about what the company’s flagship product is capable of, why the CE Mark is more important than FDA clearance and how to deal with an unpredictable regulatory process.

Hobbs
MassDevice: How did you come to join Delcath?
Eamonn Hobbs: I came to Delcath in 2008 as a new board member. The prior CEO was looking to bring significant industry experience to the board of directors as the company moved toward finishing up its Phase III clinical trial.
In 2009 I was appointed CEO by the board because the commercialization phase of the company was near and the prior CEO did not have any prior commercial experience in the medical field. It was felt it was time to bring on someone with significant experience, and I certainly fit that bill.
I’m a serial entrepreneur, a biomedical engineer. I’ve started a number of companies. The company before Delcath was AngioDynamics, which was a very successful company that I started in 1988 and took public in 2004. It had a very, very successful run of 21 years.
MassDevice: You’ve been in the industry a long time.
EH: This will be my 31st year in the med-tech and combination-product arena. I often joke that God help me if I had to do anything else, because I wouldn’t have a clue. I’m very lucky, in that I’ve been blessed with an opportunity to work in a field that I get tremendous satisfaction from and certainly know pretty well.
Not everybody looks forward to going to work every day. Helping patients and improving health care is a very potent motivator and a very gratifying one, when it works.
MassDevice: Chemosat is a different approach to cancer therapy. What’s its competition?
EH: There are no competing regional therapies for the liver. There are systemic therapies which treat the entire body, including the liver, but those therapies are typically ineffective in providing disease control in the liver.
On the opposite end of the spectrum, there are a number of focal therapies — see a tumor, treat a tumor. The gold standard in focal therapy is resection, but very few patients are good candidates for resection. Only about 10 percent of liver cancer patients are resectable. The other 90 percent have very few options.
There are a number of other focal therapies, but very often the disease is widespread in the liver and the focal therapy is only a palliative measure, to control certain tumors, to provide some symptom relief or other clinical benefits.
We, in short, are creating a whole new regional therapy space to address a very large unmet clinical need. Once cancer gets into the liver, outside of resection, which only can benefit a small minority of the patients, there aren’t any really good solutions for the patients. So we’re very excited about the Delcath technology and that large medical unmet need.
MassDevice: Chemosat cleans chemo out of the blood before it gets to the rest of the body. How clean does it get?
EH: The Phase III trial was conducted with a filtering technology that filtered approximately 70 percent of the drug out of the blood, which translated into the patient’s systemic circulation receiving the equivalent of a typical IV dose of the same drug.
The downside of that is that the systemic effects of the melphalan hydrochloride that we use is bone marrow suppression and, although a manageable complication and a very well-understood complication, it’s still a pretty significant issue.
We’ve developed a new filtering technology that removes 98 percent or more of the drug, which we believe will materially diminish the systemic effects of the now-very-tiny amount of drug that gets into the systemic circulation, which should really provide tremendous benefits for both patients and clinicians.
The systemic toxicity should be greatly reduced and the bone marrow suppression should be greatly reduced, if not made immaterial because it’s not measurable. That not only will make the post-op treatment of the patients a lot more straightforward, but it will also allow medical oncologists to continue treating their patients systemically shortly after receiving the Delcath treatment. Before, they had to wait for the bone marrow to recover before they could continue to treat with systemic chemotherapy occasions.
For cancers outside of the liver, such as virtually all metastatic cancers where the primary tumor came from an organ outside the liver, this is very important. The medical oncologists not only want to control disease in the liver, they want to keep controlling disease outside the liver. The new filtering technology, we think, is a really a breakthrough advance and we’re very excited about it.
We expect to roll that out in Europe in 2012, next year, and in the U.S. we’re negotiating with the FDA as to how we would pursue regulatory approval for the improved filter, but we don’t know the pathway quite yet.
MassDevice:
What are the next steps for Chemosat?
EH: We obtained our European approval in April of this year. We’re gearing up to start commercial operations in Europe, which we expect will start in Q4 of this year. We’re in the building-inventory phase and establishing-commercial-infrastructure phase.
The European approval is a CE Mark for a Class III medical device, as the drug that our medical device delivers is already approved and routinely used in the European Union.
We got a very broad indication in our CE Mark that covers all liver cancers. That translates into about 95 percent of the world market opportunity outside the U.S.
There’s a push-and-pull scenario here, where we’re going to create a marketing-detailing-sales organization via a contract sales organization, to the medical oncology community. They will order the procedure to be done by the interventional radiologist and then we’ll sell to and train the interventional radiologist to conduct the procedure.
The CE Mark that we have in Europe is a very important regulatory approval, because virtually every country in the world outside of the U.S. accepts the CE Mark as a predicate regulatory approval and provides reciprocal approval.
So we’ll be leveraging the CE Mark around the world to gain regulatory approval in all the emerging markets in relatively short order. We expect to have regulatory approval in countries in Asia, South America, Central America and Canada, the Middle East and Africa starting in 2012. We should have regulatory approval in virtually all the countries by the end of 2013.
Since the market opportunities are so much bigger outside the U.S., if I had to choose between the two regulatory approvals, I would take the one we got, the CE Mark, far ahead of the FDA’s, from a commercial perspective.
MassDevice: And in the U.S.?
EH: We plan to file our NDA by the end of the year, a new drug application, because our drug-device combination is regulated as a strict combination product that requires new drug labeling as well. We folded the drug development and device development pathway into the new drug approval pathway, so we’re following a new drug application.
That application will go in prior to the end of the year. That’s our intent and we would hope to have U.S. approval for a relatively narrow indication, compared to Europe, of melanoma mets [Eds. note: metastatic] to the liver. That’s why there’s such a marked differential between outside the U.S. and inside the U.S.; it’s heavily weighted towards the outside U.S. market, with 95 percent.
With the filing of our NDA by the end of the year, we would anticipate a six-month review and U.S. approval in mid-year 2012 for the indication of melanoma mets to the liver.
MassDevice: Your original NDA was refused by the FDA. What happened there?
EH: We got a refusal-to-file, an RTF, in February of this year. We filed in December. The refusal-to-file is a mechanism that the FDA uses to inform you that the application was insufficient to allow a review, which surprised us, frankly. We conducted our Phase III trial under an SPA, a special protocol assessment, which is a formal agreement with the agency on all the facts and circumstances associated with the trial.
We were quite confident we had supplied all the data that was part of the SPA to the agency. But, although they agreed we had an SPA, they felt the hospitalization data was insufficient to allow them to conduct a thorough review of the safety profile of the procedure. They wanted additional hospital data that was not collected in the original case report forms that were approved as part of the SPA.
After clarifying that with them, we’ve embarked on a monitoring program to go back to the patient records and mine hospitalization data that the FDA is specifically looking for. That’s why it’s taking us to the end of the year to reformat the application, to satisfy what the agency is looking for.
MassDevice: What changed between the original agreement and the close of the trial?
EH: I would be guessing. We certainly asked that question. The SPA was agreed to in 2005, and the FDA of 2005 is certainly not the FDA of 2011. With regards to safety, the agency is well within its rights to ask for whatever it wants.
The good news is that data is readily available. The bad news is the process of getting it is time-consuming. It basically added a year — it’ll be a year between when we filed the NDA originally and when we refile.
MassDevice: Delcath’s stock has been a roller coaster ride over the past year; you’ve presided over some pretty big spikes and troughs. What do you attribute the stock’s volatility to?
EH: Delcath is an extremely volatile stock and there a number of components to that. One is that it’s a stock that’s shareholder based, that’s dominated by retail shareholders who tend to be much more active traders. The trading volume is high and the volatility seems to be equally high. The stock seems to go up significantly on good news and down significantly on bad news — or no news.
I believe the shareholders have a lack of appreciation for how big and accessible the European and other outside-U.S. markets are for Delcath technology. The shareholder base is very U.S.-centric and U.S.-fixated. Ninety-five percent of our market opportunity happens to be outside of the U.S., which we got an approval for. We have a very significant education process to go through with the shareholders, which also includes bringing on more global shareholders. Europeans are potentially very Euro-centric. Since we’re going to be rolling out in Europe imminently, that may be of more interest to European investors than some American investors. There’s a lot of moving parts there.
MassDevice: How do you deal with the ups and downs of the market?
EH: I would love to tell you that I don’t watch the stock constantly, and I keep a long-term view constantly, because that’s the right thing to do. But contrary to popular belief, most — if not all — CEOs are human. We tend to be cut from the same cloth, in that we’re very competitive and we like to see the stock price higher rather than lower.
I’m acutely aware of the share price most days and the direction that it’s going in and, of course, every day I try and add value and try and make decisions that are going to increase the shareholders’ value in the company. But most of the decisions that any company executive can make are such that they’re not going to increase the value that day, or that month, or maybe even that year. They’re much more strategic in effect, so there’s a bit of a lag between what we do today and the benefits of it being appreciated in the stock price.
MassDevice: In addition to your role as Delcath CEO, you’re also the sitting chairman of the board for the Medical Device Manufacturers’ Assn. What are some of your concerns about the current regulatory environment in the U.S.?
EH: As an industry, we’re very concerned about the current regulatory environment and are looking to work with the FDA and legislators to improve that situation. The biggest issue we face is the lack of transparency and predictability in the regulatory process in the U.S. That has really happened over the last six years.
What’s interesting about that situation is that the statute hasn’t changed during that period. The same law is still in effect, but the regulatory environment has definitely decayed to a very unfavorable one for new innovations to reach patients in the U.S. in a timely fashion. The issue is an extremely important one, in that we as a country really need to get innovation to patients in a timely fashion and not lose jobs in the industry to Europe and other countries based on much more predictable regulatory pathways being available.
It’s a big issue for everyone in the industry and we’re working very, very hard to collaborate with the agency and with our legislators to figure out how to fix it.
It’s not a problem you can put your finger on and say there’s one magic bullet to fix it. Clearly there is a situation where we had a much more productive regulatory pathway in 2005 and in 2011 we have a very unproductive regulatory pathway. In solving a problem, first you have to identify what the problem is. We know the law didn’t change, so something else did.
